Tổng quan về cục diện kháng thuốc EGFR TKI trong ung thư phổi không tế bào nhỏ có đột biến EGFR: Cơ chế và các hướng tiếp cận điều trị mới
BSNT. Nguyễn Thị Thuý Hường2, GS. TS. Mai Trọng Khoa1,2, PGS. TS. Phạm Cẩm Phương1,2, PGS. TS. Phạm Văn Thái1,3,
(1) Trung tâm Y học hạt nhân và Ung bướu - Bệnh viện Bạch Mai
(2) Trường Đại học Y Dược - Đại học Quốc gia Hà Nội
(3) Trường Đại học Y Hà Nội
(Bài dịch)
TÓM TẮT
Sự kháng EGFRTKI (Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitor – thuốc ức chế tyrosine kinase yếu tố phát triển nội mô) vẫn là thách thức lớn trong điều trị bệnh nhân ung thư phổi không tế bào nhỏ có đột biến EGFR. Mặc dù các thế hệ TKI mang lại hiệu quả điều trị mang tính đột phá, từ TKI thế hệ đầu tiên ức chế hồi phục như erlotinib và gefitinib cho đến thế hệ ba tiêu chuẩn điều trị hiện tại ức chế cộng hoá trị, kháng thuốc nguyên phát hoặc mắc phải đối với các thuốc này vẫn xuất hiện tất yếu thông qua nhiều cơ chế khác nhau. Sự xuất hiện của các liệu pháp điều trị kết hợp phổi hợp hoá trị, ức chế tăng sinh mạch và kháng thể đặc hiệu kép hoặc liên hợp kháng thể - thuốc đã gia tăng lợi ích lâm sàng nhưng đồng thời cũng tạo ra các kiểu hình kháng thuốc mới, nhấn mạnh tính linh hoạt và phức tạp trong tiến hóa của khối u dưới áp lực điều trị. Trong bài tổng quan này, chúng tôi cung cấp một tổng hợp toàn diện về các cơ chế phân tử gây kháng thuốc đối với EGFR TKIs thế hệ thứ ba, mô tả các chiến lược điều trị dựa trên biomarker (dấu ấn sinh học) hoặc không dựa trên biomarker nhằm vượt qua các cơ chế này, và thảo luận về những cách tiếp cận mới nổi để ngăn ngừa kháng thuốc thông qua việc áp dụng sớm các liệu pháp kết hợp. Chúng tôi nhấn mạnh sự chuyển dịch mô hình từ theo dõi bằng hình ảnh học sang theo dõi bằng phân tử đối với tình trạng kháng thuốc, đồng thời khám phá cách mà những tiến bộ trong phân tích DNA khối u lưu hành, trí tuệ nhân tạo và đa omics (nghiên cứu nhiều loại sinh học phân tử) có thể hỗ trợ các chiến lược điều trị thích ứng. Khi bối cảnh điều trị tiếp tục phát triển, việc hiểu rõ hơn về cơ chế kháng thuốc sẽ trở nên thiết yếu nhằm định hướng hợp lý cho việc sắp xếp trình tự điều trị, cung cấp thông tin cho thiết kế thử nghiệm lâm sàng và cải thiện kết quả lâu dài cho bệnh nhân ung thư phổi không tế bào nhỏ (NSCLC – non small cell lung cancer) có đột biến EGFR.
GIỚI THIỆU
Các thuốc ức chế tyrosine kinase của EGFR (EGFR TKIs) đã tạo ra cuộc cách mạng trong mô hình điều trị cho bệnh nhân ung thư phổi không tế bào nhỏ (NSCLC) giai đoạn tiến xa có đột biến EGFR, giúp cải thiện đáng kể tiên lượng và chất lượng cuộc sống.Tuy nhiên, tình trạng kháng thuốc đối với các thuốc này và sự tiến triển bệnh vẫn là điều tất yếu, đặt ra thách thức lâm sàng thường trực. Kháng EGFR TKI xuất hiện thông qua sự chọn lọc dòng của tế bào ung thư dưới áp lực tiến hóa về không gian và thời gian, đồng thời được thúc đẩy bởi các biến đổi gen mắc phải.Hiện tượng kháng này được phân loại thành kháng nguyên phát (nội tại) hoặc kháng thứ phát (mắc phải). Kháng nguyên phát thường được định nghĩa là sự tiến triển bệnh xảy ra trong vòng 3 tháng sau khi bắt đầu EGFR TKI hoặc khi đáp ứng điều trị tốt nhất vẫn là bệnh tiến triển. Hiện tượng này chủ yếu liên quan đến sự dị thể cấu trúc của các dạng đột biến EGFR khác nhau, ảnh hưởng đến khả năng gắn kết của TKI, hoặc sự đồng tồn tại của các oncogene khác duy trì sự sống và tăng sinh của tế bào độc lập với EGFR. Kháng thứ phát đề cập đến sự tiến triển bệnh xảy ra sau một đáp ứng điều trị ban đầu hiệu quả (đáp ứng khách quan hoặc bệnh ổn định) kéo dài hơn 6 tháng. Kháng mắc phải này có thể được phân loại thêm thành kháng phụ thuộc EGFR (on-target), kháng không phụ thuộc EGFR (off-target) hoặc chuyển dạng mô học. Tuy nhiên, các cơ chế nền tảng của kháng nguyên phát và thứ phát vẫn phần lớn chưa được biết rõ, với khoảng 50% trường hợp kháng mắc phải đối với osimertinib hàng đầu không được giải thích bằng các biến đổi đơn gen đã biết. Điều này không chỉ làm nổi bật sự phức tạp của các cơ chế thoát khỏi điều trị của khối u, mà còn nhấn mạnh tầm quan trọng của các nghiên cứu toàn diện, đặc biệt là sự cần thiết của các cách tiếp cận đa-sinh học phân tử (multi-omic) — bao gồm nhưng không giới hạn ở biểu sinh học (epigenomics), phiên mã học (transcriptomics), proteomics và metabolomics —nhằm xác định các cơ chế kháng thuốc mới vượt ngoài phạm vi của phân tích bộ gen đơn thuần.
Cách tiếp cận trong việc theo dõi tình trạng kháng điều trị đã chuyển dịch từ ‘kháng thuốc qua hình ảnh học’ truyền thống, vốn dựa trên đo lường bằng chẩn đoán hình ảnh,sang ‘kháng thuốc qua phân tử’, dựa trên đánh giá động học của DNA khối u lưu hành (ctDNA). Đặc biệt, sự thay đổi này đã cho phép phát hiện và theo dõi bệnh tồn dư tối thiểu (MRD) ở những bệnh nhân không có tổn thương nhìn thấy trên hình ảnh, từ đó nhận diện sớm tình trạng kháng thuốc. Tuy nhiên, việc lập hồ sơ phân tử theo dõi tiến cứu và nghiên cứu cắt dọc vẫn gặp nhiều thách thức, gồm hạn chế về mẫu mô để phát triển các bảng ctDNA cá thể hóa theo khối u cho một số bệnh nhân, kết quả âm tính giả phụ thuộc vào gánh nặng khối u và thiếu dữ liệu liên quan đến chuyển dạng mô học, cũng như các vấn đề kinh tế – dược. Đáng chú ý, chuyển dạng mô học từ NSCLC có đột biến EGFR sang ung thư phổi tế bào nhỏ (SCLC) là một cơ chế quan trọng gây kháng EGFR TKIs, xảy ra ở khoảng 3–10%bệnh nhân phát triển kháng mắc phải theo các nghiên cứu sinh thiết lặp lại.Những tiến bộ trong đa omics đã mang lại hiểu biết sâu hơn về các cơ chế nền tảng của quá trình chuyển dạng sang tế bào nhỏ, làm rõ các protein và con đường tín hiệu chính liên quan, đồng thời đặt nền tảng lý thuyết cho việc phát triển các chiến lược điều trị mới.
Các chiến lược hiện tại nhằm giải quyết tình trạng kháng EGFR TKIs chủ yếu tập trung vào hai hướng tiếp cận: vượt qua tình trạng kháng đã hình thành hoặc trì hoãn sự khởi phát của nó. Các chiến lược nhằm vượt qua kháng thuốc đã hình thành có thể được phân loại thành tiếp cận dựa trên dấu ấn sinh học và không dựa trên dấu ấn sinh học, tùy thuộc vào sự hiện diện hoặc vắng mặt của các cơ chế kháng thuốc cụ thể vốn tạo ra độ nhạy bổ sung đối với các liệu pháp nhắm trúng đích phân tử khác.Các tiếp cận dựa trên dấu ấn sinh học đòi hỏi phải có hồ sơ phân tử toàn diện,điều này có thể hạn chế khả năng tiếp cận hoặc làm chậm quá trình điều trị,nhưng có thể được hỗ trợ bởi sự chuyển dịch đã đề cập trước đó sang phân tích ctDNA. Ngược lại, các chiến lược nhằm trì hoãn kháng thuốc thường không yêu cầu quản lý cá thể hóa như vậy và thường bao gồm việc kết hợp EGFR TKIs ngay từ đầu với các liệu pháp khác nhằm loại bỏ các tế bào dung nạp thuốc (DTPs) và/hoặc nhắm trúng các con đường kháng thuốc một cách chủ động, với mục tiêu kéo dài thời gian đáp ứng (DOR). Việc xác định liệu các liệu pháp kết hợp mới này có làm thay đổi các mô hình kháng thuốc truyền thống hay có thể tạo ra các cơ chế kháng thuốc mới là một lĩnh vực nghiên cứu đang nổi lên trong tương lai. Các thuốc EGFR TKIs thế hệ thứ ba như osimertinib đã trở thành tiêu chuẩn điều trị (SOC) hàng đầu cho bệnh nhân ung thư phổi không tế bào nhỏ (NSCLC) giai đoạn tiến xa có đột biến EGFR. Các cơ chế kháng thuốc chính đối với EGFR TKIs thế hệ trước đã được xác định rõ, trong đó đột biến EGFR T790M tại vị trí đích — khiến EGFR vẫn nhạy cảm với thuốc ức chế thế hệ thứ ba — chiếm khoảng 50% trường hợp.Do đó, bài tổng quan này tập trung vào việc hệ thống hóa những tiến bộ liên quan đến cơ chế và quản lý tình trạng kháng EGFR TKIs thế hệ thứ ba. Thông qua thảo luận chuyên sâu về các chủ đề này, chúng tôi cung cấp một nguồn tài liệu giá trị và định hướng cho nghiên cứu cũng như thực hành lâm sàng trong lĩnh vực này.
1. Cơ chế kháng EGFR
1.1. Kháng nguyên phát đối với đơn trị liệu
Kháng nguyên phát đối với đơn trị liệu bằng EGFR TKI chủ yếu được cho là do thiếu sự phụ thuộc vào đích tác động, dẫn đến bệnh tái phát nhanh chóng sau khi bắt đầu điều trị, mặc dù hiện nay định nghĩa lâm sàng về hiện tượng kháng nội tại này vẫn chưa được thiết lập rõ ràng. Kobayashi và cộng sự đã đề xuất hai dạng kháng nguyên phát: ‘kháng nguyên phát nội tại’, được định nghĩa là hoàn toàn không có đáp ứng với điều trị hàng đầu bằng EGFR TKI (tức là đáp ứng tốt nhất vẫn là bệnh tiến triển); và ‘kháng nguyên phát muộn’, đặc trưng bởi tái phát sớm trong vòng 6 tháng sau khi có đáp ứng hình ảnh ban đầu (bệnh ổn định hoặc thậm chí có đáp ứng khách quan). Tuy nhiên, một số nghiên cứu lâm sàng, bao gồm cả thử nghiệm tiến cứu và nghiên cứu dịch chuyển, đã áp dụng ngưỡng nghiêm ngặt hơn là 3 tháng, đặc biệt khi bệnh tiến triển là đáp ứng tốt nhất được ghi nhận.
Kháng nguyên phát đối với osimertinib xảy ra ở khoảng 4–10% bệnh nhân UTPKTBN giai đoạn tiến xa có đột biến EGFR. Các cơ chế nền tảng của hiện tượng kháng nguyên phát này chưa được làm sáng tỏ hoàn toàn, mặc dù những nguyên nhân đã được xác định bao gồm sự dị thể cấu trúc của các đột biến EGFR và các biến đổi gen đồng xuất hiện trong những con đường sinh ung khác. Ví dụ, cấu hình không gian đặc thù của các đột biến chèn exon 20 EGFR dẫn đến giảm ái lực với các EGFR TKIs thông thường,bao gồm cả osimertinib. Các thuốc TKI mới đã được phát triển để nhắm vào các đột biến chèn exon 20 EGFR, đã được tổng quan chi tiết trước đây nhưng nằm ngoài phạm vi của bài viết này. Các nghiên cứu đoàn hệ hồi cứu đã cho thấy rằng khuếch đại MET de novo, khuếch đại HER2 và/hoặc các biến đổi gen thuộc con đường bù trừ khác cũng liên quan đến kháng nguyên phát đối với EGFR TKIs thế hệ thứ ba. Đột biến TP53, xuất hiện ở 30–50% NSCLC có đột biến EGFR, có thể thúc đẩy nhanh tình trạng kháng EGFR TKIs thông qua hoạt hóa kéo dài ERK và MYC. Các đột biến đồng tồn tại trong các gen liên quan đến chu kỳ tế bào (ví dụ CCND1/2, CCNE1 và CDK4/6), hoặc thiếu hụt PTEN và/hoặc đột biến PIK3CA làm gián đoạn sự ức chế con đường PI3K–AKT ở hạ lưu EGFR, cũng góp phần vào kháng nội tại đối với EGFR TKIs. Ngoài ra, một đa hình mất đoạn mầm ở intron 2 của gen mã hóa BIM, xuất hiện ở khoảng 13% người Đông Á, đã được chứng minh làm giảm hoạt tính của protein tiền chết theo chương trình quan trọng này và do đó làm giảm độ nhạy với osimertinib.
Các cơ chế đa dạng nền tảng của tình trạng kháng nguyên phát với EGFR TKIs nhấn mạnh nhu cầu cần có các chiến lược điều trị cá thể hóa dựa trên phân tích phân tử rộng hơn ngay từ khi chẩn đoán. Bằng chứng cho thấy những bệnh nhân được xác định có các cơ chế kháng nguyên phát tiềm tàng có thể hưởng lợi từ các liệu pháp kết hợp nhắm vào các yếu tố sinh ung bù trừ (ví dụ như MET TKIs cho những trường hợp có đồng biến đổi MET), nhằm ức chế các con đường kháng thuốc ở giai đoạn sớm, từ đó trì hoãn tiến triển bệnh và cải thiện kết quả sống còn. Chiến lược này sẽ được thảo luận chi tiết hơn trong phần tiếp theo của bài tổng quan.
1.2. Kháng thứ phát đối với đơn trị liệu
Kháng thứ phát (mắc phải) đối với EGFR TKIs liên quan đến sự xuất hiện tình trạng kháng thuốc sau khi có đáp ứng trên hình ảnh và/hoặc lợi ích lâm sàng duy trì (kéo dài ít nhất 3 tháng và thường hơn 6 tháng). Kháng mắc phải thường được phân loại theo cơ chế sinh học nền tảng, như kháng phụ thuộc EGFR (on-target), kháng không phụ thuộc EGFR (off-target) hoặc chuyển dạng mô học. Nhìn chung, các mô hình kháng đối với osimertinib bước một tương tự như những gì được báo cáo với osimertinib bước hai. Tuy nhiên, so với các EGFR TKIs thế hệ trước (như erlotinib và gefitinib), việc sử dụng osimertinib ngay từ đầu có liên quan đến tỷ lệ thấp hơn của kháng phụ thuộc EGFR tại vị trí đích (ví dụ đột biến EGFRC797X) và tỷ lệ cao hơn của các cơ chế chưa rõ (có khả năng là off-target).
Kháng phụ thuộc EGFR (on-target) đối với EGFR TKIs thế hệ thứ ba chủ yếu biểu hiện dưới dạng các đột biến thứ phát của EGFR. Theo một phân tích trên mẫu ctDNA huyết tương từ bệnh nhân được điều trị bằng osimertinib trong thử nghiệm phaIII FLAURA, đột biến C797S tại exon 20 là phổ biến nhất (6%), tiếp theo là L718Q (2%), G796R/D (1%), S768I (1%) và một số đột biến khác. Những đột biến này làm giảm độ nhạy của EGFR đối với EGFR TKIs bằng cách ngăn cản sự gắn kết của thuốc hoặc làm giảm đáng kể ái lực gắn kết, từ đó dẫn đến tình trạng kháng mắc phải. Đặc biệt, đột biến C797S ngăn cản sự gắn kết cộng hóa trị của các EGFR TKIs không hồi phục như osimertinib tại vị trí này.
Kháng phụ thuộc EGFR (on-target) đối với EGFR TKIs thế hệ thứ ba chủ yếu biểu hiện dưới dạng các đột biến thứ phát của EGFR. Theo một phân tích trên mẫu ctDNA huyết tương từ bệnh nhân được điều trị bằng osimertinib trong thử nghiệm phaIII FLAURA, đột biến C797S tại exon 20 là phổ biến nhất (6%), tiếp theo là L718Q (2%), G796R/D (1%), S768I (1%) và một số đột biến khác. Những đột biến này làm giảm độ nhạy của EGFR đối với EGFR TKIs bằng cách ngăn cản sự gắn kết của thuốc hoặc làm giảm đáng kể ái lực gắn kết, từ đó dẫn đến tình trạng kháng mắc phải. Đặc biệt, đột biến C797S ngăn cản sự gắn kết cộng hóa trị của các EGFR TKIs không hồi phục như osimertinib tại vị trí này.
Các cơ chế kháng thuốc ngoài EGFR (off-target) bao gồm khuếch đại MET, biến đổi HER2 và đột biến BRAF và/hoặc KRAS, trong đó phổ biến nhất là khuếch đại MET (tỷ lệ >15% sau điều trị hàng đầu bằng osimertinib), vốn bỏ qua EGFR để hoạt hóa các con đường tín hiệu phía dưới STAT, MAPK và PI3K. Các dung hợp gen thụ thể tyrosine kinase (RTK), chủ yếu là RET, ALK, BRAF hoặc FGFR, cũng là một cơ chế kháng off-target thường gặp (tỷ lệ 3–5%). Trong tổng quan tài liệu trước đây của chúng tôi công bố năm 2024, đã xác định tổng cộng 291 bệnh nhân có dung hợp gen RTK được báo cáo là cơ chế kháng mắc phải sau điều trị bằng EGFR TKI trong 59 nghiên cứu.Trong nhóm này, có 300 gen dung hợp khác nhau được ghi nhận, phổ biến nhất là RET (chiếm 50% tổng số dung hợp RTK), tiếp theo là BRAF (13,3%), ALK (13,3%),FGFR (10,0%), NTRK (5,3%), EGFR (1,7%), ROS1 (1,3%), MET (1,0%) và ERBB3/4 (0,7%). Ngoài ra, chúng tôi hồi cứu thêm 27 bệnh nhân tại năm trung tâm y tế,những người đã được điều trị NSCLC có đột biến EGFR và phát hiện dung hợp gen mắc phải bằng giải trình tự thế hệ mới (NGS) từ mẫu mô hoặc máu sau khi bệnh tiến triển trên EGFR TKI (trong đó 16/27, 59,3% dùng osimertinib); dung hợp ALK(12/27, 42,9%) và dung hợp RET (10/27, 35,7%) là phổ biến nhất. Việc xác định dung hợp RTK có ý nghĩa quan trọng vì chúng có tiềm năng trở thành mục tiêu điều trị. Các biến đổi dẫn đến hoạt hóa các con đường tín hiệu hạ lưu của EGFR cũng là cơ chế kháng off-target quan trọng. Ví dụ được xác định ở bệnh nhân dùng osimertinib bao gồm đột biến và/hoặc khuếch đại PIK3CA (3–7%), các biến đổi tái hoạt hóa con đường RAS–MAPK (3–5%) như đột biến KRAS hoặc BRAF, và các bất thường gen liên quan đến chu kỳ tế bào (10% trong điều trị bước một và 15%trong điều trị bước hai), như liên quan đến CCND1/D2/E1, CDK4/6 và/hoặc CDKN2A.
Chuyển dạng mô học và tính dẻo tuyến tính xảy ra ở khoảng 12–15% bệnh nhân tiến triển khi điều trị bằng EGFR TKIs. Ung thư phổi tế bào nhỏ (SCLC) là dạng mô học chuyển dạng phổ biến nhất (3–10%), ngoài ra còn có ung thư biểu mô vảy (cần lưu ý rằng NSCLC có đột biến EGFR thường là ung thư biểu mô tuyến không vảy) và ung thư dạng sarcomatoid. Sự hiện diện đồng thời của đột biến RB1 và TP53 cùng với đột biến EGFR trước điều trị có liên quan đến nguy cơ tăng chuyển dạng sang SCLC,với 25% bệnh nhân biểu hiện thành phần SCLC ngay từ đầu hoặc cuối cùng có chuyển dạng sang tế bào nhỏ. Một nghiên cứu hồi cứu trên 25 bệnh nhân có chuyển dạng từ NSCLC sang SCLC sau điều trị bằng EGFR TKI cho thấy các đột biến đồng xuất hiện RB1, TP53 và PIK3CA lần lượt ở 68%, 36% và 12% sau khi chuyển dạng sang SCLC. Một phân tích khác trên 65 bệnh nhân ung thư phổi biểu mô tuyến có đột biến EGFR cho thấy mất chức năng đồng thời của RB1 và TP53 trước điều trị bằng EGFR TKI làm tăng nguy cơ chuyển dạng sang SCLC hơn 40 lần (RR 42,8; KTC95%: 5,88–311).
Các nhà nghiên cứu đã phát triển mô hình chuột biến đổi gen để khảo sát đặc điểm phiên mã ở các giai đoạn khác nhau của quá trình chuyển dạng từ ung thư biểu mô tuyến phổi mang đột biến EGFR sang ung thư phổi tế bào nhỏ (SCLC). Họ phát hiện rằng MYC điều khiển chương trình phiên mã của SCLC, trong khi EGFR điều khiển ung thư biểu mô tuyến, với một trạng thái trung gian giống tế bào gốc được quan sát thấy trong quá trình chuyển dạng. Như vậy, MYC thúc đẩy sự biệt hóa của các tế bào giống tế bào gốc này theo hướng SCLC, trong khi mất RB1 được xác định là cần thiết nhưng không đủ để gây chuyển dạng. Nghiên cứu này làm nổi bật một cơ chế mới của chuyển dạng sang tế bào nhỏ, đồng thời hỗ trợ các chiến lược điều trị nhắm vào MYC.
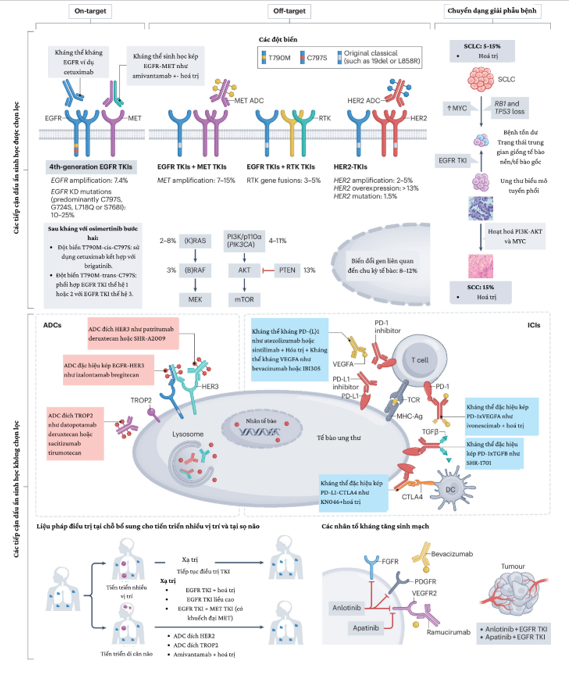
Hình 1 | Các cơ chế kháng mắc phải đối với EGFR TKIs thế hệ thứ ba và chiến lược quản lý tương ứng. Kháng thuốc thứ phát (secondary resistance) đối với các thuốc ức chế tyrosine kinase EGFR (EGFR TKIs) thế hệ thứ ba có thể phát sinh thông qua nhiều cơ chế khác nhau, gồm: kháng on-target do các biến đổi bổ sung trong EGFR; kháng off-target do các bất thường trong các con đường tín hiệu thụ thể tyrosine kinase (RTK) thay thế và/hoặc các thành phần ở phía dưới của con đường tín hiệu EGFR; và chuyển dạng mô học sang một dòng tế bào khác vốn có tính kháng thuốc nội tại.Khi xác định được cơ chế cụ thể, có thể áp dụng các chiến lược điều trị phù hợp được lựa chọn dựa trên biomarker. Trong trường hợp không xác định được cơ chế,tình trạng kháng EGFR TKI có thể được xử lý theo hướng rộng hơn bằng các phương pháp điều trị không dựa trên biomarker, nhắm vào các đặc điểm chung của khối u kháng thuốc, như kháng nguyên khối u phổ biến, vi môi trường khối u ức chế miễn dịch và/hoặc hệ mạch máu khối u (tân sinh mạch). Các phương pháp không dựa trên biomarker cũng gồm điều trị tại chỗ như xạ trị triệt để định vị (stereotactic ablative radiotherapy) hoặc phẫu thuật, có thể được sử dụng để loại bỏ bệnh tồn dư ít ổ, bệnh tiến triển ít ổ và/hoặc bệnh tại hệ thần kinh trung ương (CNS),đồng thời tiếp tục duy trì EGFR TKI đơn độc hoặc kết hợp với các liệu pháp toànthân khác (dựa trên biomarker hoặc không). Các thuật ngữ viết tắt: ADC: kháng thể–thuốc liên hợp (antibody–drug conjugate) DC: tế bào tua (dendriticcell) ICI: thuốc ức chế điểm kiểm soát miễn dịch (immune-checkpoint inhibitor)KD: vùng kinase (kinase domain) MHC–Ag: phức hợp hòa hợp mô chính – trình diện kháng nguyên SCC: ung thư biểu mô vảy (squamous cell carcinoma) SCLC: ung thư phổi tế bào nhỏ (small-cell lung cancer) TCR: thụ thể tế bào T (T cellreceptor).
Cơ chế kháng thuốc đối với liệu pháp kết hợp dựa trên EGFR TKI Hiện nay, các chiến lược điều trị kết hợp đã được thiết lập hoặc đang được nghiên cứu dành cho NSCLC có đột biến EGFR gồm: EGFR TKIs kết hợp với hóa trị, kháng thể đặc hiệu kép, thuốc kháng tăng sinh mạch, liên hợp thuốc kháng thể (ADC) hoặc các loại TKI khác. Cho đến nay, các phác đồ kết hợp như osimertinib với hóa trị kép nền platinum và lazertinib với kháng thể hai đặc hiệu EGFR–MET amivantamab đã được phê duyệt trong điều trị đầu tay. Những chiến lược kết hợp này có thể cải thiện thời gian sống không bệnh tiến triển (PFS) và thời gian sống toàn bộ (OS), tuy nhiên cũng có thể dẫn đến sự xuất hiện của các kiểu kháng thuốc hoặc cơ chế kháng mới.
Trong thử nghiệm then chốt pha III FLAURA2 đánh giá liệu pháp hàng đầu osimertinib kết hợp hóa trị, các cơ chế kháng thuốc đã được phân tích bằng ctDNA. Kết quả cho thấy bệnh nhân nhận phác đồ kết hợp này có tỷ lệ mắc phải đột biến EGFR C797S thấp hơn (4% so với 12% ở nhóm dùng osimertinib đơn trị) và khuếch đại MET thấp hơn (9% so với 14%), nhưng lại có sự gia tăng đáng kể tỷ lệ bệnh nhân mang các đột biến kháng mắc phải chưa rõ cơ chế (75% so với 49%).
Sự kết hợp lazertinib và amivantamab đã được phê duyệt trong điều trị hàng đầu dựa trên dữ liệu từ thử nghiệm pha III MARIPOSA. Phân tích thăm dò dựa trên NGS của ctDNA cho thấy, so với đơn trị liệu bằng osimertinib, bệnh nhân dùng amivantamab kết hợp lazertinib có tỷ lệ mắc phải khuếch đại MET thấp hơn rõ rệt (4,4% so với 13,6%;P = 0,017) và tỷ lệ đột biến EGFR thứ phát tại vị trí đích (bao gồm C797S, L718X và G724X) cũng thấp hơn (0,9% so với 7,9%; P = 0,014). Xu hướng giảm tỷlệ mắc phải đột biến kháng TP53 (9,7% so với 12,9%) và mất đồng thời TP53 và RB1 (0,9% so với 2,9%) — dấu hiệu của chuyển dạng sang ung thư phổi tế bào nhỏ— cũng được ghi nhận ở nhóm amivantamab kết hợp lazertinib. Tuy nhiên, do cả hai nghiên cứu đều sử dụng sinh thiết lỏng để phân tích bộ gen tại thời điểm ban đầu và khi xuất hiện kháng thuốc, dữ liệu báo cáo có thể chỉ phản ánh một phần hạn chế của bức tranh kháng thuốc, bởi hiện tượng ctDNA tiết ra thấp và/hoặc khả năng phát hiện các đột biến tần suất thấp còn hạn chế. Do đó, cần có thêm các nghiên cứu để làm rõ cơ chế kháng thuốc ở bệnh nhân được điều trị bằng các liệu pháp kết hợp mới này cũng như các phác đồ khác.
Các nghiên cứu nêu trên đã chứng minh rõ ràng rằng liệu pháp kết hợp hàng đầu giúp kéo dài thời gian đáp ứng (DOR), nhưng tình trạng kháng thuốc gần như vẫn không thể tránh khỏi, và các cơ chế nền tảng phần lớn vẫn chưa được biết đến. Bên cạnh sự tiến hóa của các dòng tế bào ung thư, bằng chứng tiền lâm sàng và dịch chuyển gần đây cho thấy vi môi trường khối u (TME) đóng vai trò tích cực trong việc tái định hình đáp ứng điều trị và kháng thuốc. Ví dụ, một nghiên cứu giải trình tự RNA đơn bào trên mẫu sinh thiết từ 34 bệnh nhân (23 trước điều trị và 11 sau điều trị) cho thấy EGFR TKIs dẫn đến sự gia tăng số lượng đại thực bào SLC40A1+, vốn thúc đẩy sự biệt hóa tế bào T điều hòa thông qua trục CCL18–CCR8. Sự tái cấu trúc TME này có thể làm tăng tình trạng ức chế miễn dịch và từ đó tạo điều kiện cho sự phát triển kháng thuốc.
Mặc dù dữ liệu này còn sơ bộ,chúng có thể định hướng cho các nghiên cứu trong tương lai nhằm làm rõ cơ chế kháng thuốc đối với liệu pháp dựa trên EGFR TKI. Những phát hiện này cũng nhấn mạnh sự phức tạp của cơ chế kháng thuốc — vượt xa so với dự đoán ban đầu — vàcho thấy hạn chế của các công nghệ hiện tại trong việc giải thích đầy đủ sự xuất hiện của kháng thuốc. Do đó, việc sử dụng các công nghệ omics tiên tiến và cách tiếp cận đa-omics (multi-omics - phương pháp tiếp cận tích hợp dữ liệu từ nhiều ngành "omics" khác nhau (như genomics, transcriptomics, proteomics, metabolomics) là cần thiết để khám phá các cơ chế mới và cung cấp cái nhìn toàn diện hơn về kháng EGFR TKI.
2. Chuyển đổi mô hình từ kháng thuốc dựa trên hình ảnh sang kháng thuốc phân tử
Với sự phát triển của các phương pháp sinh thiết lỏng, khái niệm về “kháng thuốc” đã có sự tiến hóa đáng kể. Trước đây, kháng thuốc chủ yếu được đánh giá dựa trên chẩn đoán hình ảnh. Tuy nhiên, hiện nay, kháng thuốc cũng có thể được theo dõi thông qua sinh thiết lỏng ctDNA hoặc tế bào khối u tuần hoàn trong máu ngoại vi, từ đó đưa ra khái niệm mới về“kháng thuốc phân tử”. Những thay đổi động trong ctDNA hoặc mức tế bào khối u tuần hoàn có thể phản ánh sự biến đổi gánh nặng khối u và hồ sơ bộ gen. Trong các phân tích hồi cứu thăm dò trên mẫu máu từ các thử nghiệm FLAURA và AURA3 (đánh giá osimertinib ở first-line và second-line) tiến triển bệnh dựa trên ctDNA được quan sát thấy xảy ra trước hoặc đồng thời với tiến triển trên hình ảnh ở 56–64% bệnh nhân, với thời gian trung vị đi trước từ 1,5–3,4 tháng.
Đáng chú ý, thử nghiệm pha II APPLE đã phân ngẫu nhiên bệnh nhân NSCLC giai đoạn tiến xa có đột biến EGFR vào ba nhóm: nhóm A: dùng osimertinib cho đến khi bệnh tiến triển; nhóm B: dùng gefitinib cho đến khi phát hiện đột biến kháng EGFRT790M trong ctDNA hoặc tiếntriển trên hình ảnh theo RECIST, sau đó chuyển sang osimertinib, nhóm C: dùng gefitinib cho đến khi bệnh tiến triển theo RECIST, sau đó chuyển sang osimertinib. Trong nhóm B, chỉ 17% bệnh nhân (8/47) chuyển sang osimertinib trước khi bệnh tiến triển theo RECIST dựa trên kháng thuốc phân tử (tức là phát hiện EGFRT790M trong ctDNA), và thời gian sống không bệnh tiến triển (PFS)trung vị khi dùng osimertinib hàng hai tương tự giữa nhóm B (22,0 tháng) và nhóm C (20,2 tháng). Tuy nhiên, một hạn chế lớn của nghiên cứu này là việc sử dụng xét nghiệm real-time PCR để phân tích ctDNA, vốn có độ nhạy thấp hơn so với các phương pháp hiện đại như droplet digital PCR và giải trình tự thế hệ mới (NGS). Mặc dù còn nhiều hạn chế, thử nghiệm APPLE đã nhấn mạnh tiềm năng của việc theo dõi bệnh ở mức phân tử, không chỉ để đánh giá sự hiện diện của kháng thuốc mà còn như một công cụ hỗ trợ ra quyết định điều trị. Một thử nghiệm khả thi cho thấy việc giảm liều hoặc ngắt quãng điều trị TKI dựa trên ctDNA có thể phù hợp cho bệnh nhân NSCLC không còn tổn thương phát hiện được trên hình ảnh sau điều trị TKI đầu tay kết hợp với liệu pháp củng cố tại chỗ. Các thử nghiệm đang diễn ra, như LOCAL/CTONG1602 (NCT03046316) và APPROACH/CTONG2101 (NCT04841811), tiếp tục đánh giá tính khả thi của việc áp dụng “nghỉ thuốc” (drug holidays) dựa trên ctDNA ở bệnh nhân NSCLC, gồm cả những người có đột biến EGFR. Tuy nhiên, khả năng tiếp cận mẫu sinh thiết lại và độ nhạy của xét nghiệm vẫn là thách thức lớn đối với việc lập hồ sơ phân tử lặp lại trong thực hành lâm sàng. Trong nghiên cứu lập hồ sơ phân tử ELIOS, chỉcó 39% bệnh nhân tiến triển trên osimertinib hàng đầu thu được thành công cả mẫu khối u trước và sau điều trị.
Sinh thiết lỏng, với ưu điểm ít xâm lấn, có thể cải thiện khả năng tiếp cận, giải quyết vấn đề dị hợp khối u và phản ánh tốt hơn sinh học bệnh tổng thể. Tuy nhiên, phương pháp này cũng có những hạn chế vốn có: một tỷ lệ bệnh nhân có khối u “không tiết” ctDNA và cơ chế sinh học của việc giải phóng ctDNA vẫn chưa được hiểu rõ.
3. Chiến lược dựa trên biomarker để vượt qua kháng EGFR TKI
Các chiến lược nhằm vượt qua tình trạng kháng EGFR TKIs chủ yếu liên quan đến điều chỉnh điều trị tiếp theo dựa trên việc xác định cơ chế nền tảng, và có thể được phân loại rộng rãi thành các cách tiếp cận dựa trên biomarker hoặc không dựa trên biomarker. Các cách tiếp cận dựa trên biomarker có thể được chia nhỏ thành những chiến lược nhắm trực tiếp vào EGFRđể vượt qua kháng on-target và những chiến lược nhắm vào các yếu tố trung gian chính của kháng off-target.
Vượt qua kháng thuốc nhắm đích on-target
EGFR TKIs thế hệ thứ tư: Các thuốc ức chế EGFR chọn lọc đột biến thế hệ thứ tư gồm chất ức chế allosteric và chất ức chế cạnh tranh ATP, chủ yếu nhắm vào EGFR mang đột biến C797S. Chất ức chế allosteric EGFR: bao gồm EAI045, JBJ-04-125-02 và JBJ-09-063. Các thuốc này tác động bằng cách gắn ngoài túi gắn ATP và ức chế hoạt tính kinase của EGFR thông qua việc ổn định cấu hình bất hoạt “C-helix out”. Chất ức chế cạnh tranh ATP: được thiết kế để chọn lọc các dạng EGFR đột biến, gồm BLU-945, BBT-176, silevertinib (BDTX-1535), TQB3804, H002, BI-4020, THE-349 và JND3229. Hầu hết đang ở giai đoạn tiền lâm sàng hoặc thử nghiệm lâm sàng sớm.
Kết hợp các thuốc nhắm EGFR: Việc kết hợp nhiều thuốc nhắm EGFR là một chiến lược điều trị khả thi nhằm vượt qua tình trạng kháng on-target đối với EGFR TKIs. Ở những bệnh nhân có cả đột biến T790M và C797S sau điều trị bằng EGFR TKIs thế hệ thứ nhất và thứ ba, việc phân biệt cấu hình cis và trans của các đột biến đồng thời này là rất quan trọng,trong đó cấu hình cis (cả hai đột biến xảy ra trên cùng một alen EGFR) phổ biến hơn, chiếm khoảng 82% trường hợp. Sự phân biệt này không liên quan đến bệnh nhân kháng thuốc với EGFR TKIs thế hệ ba khi điều trị đầu tay, bởi đột biến T790M không gây kháng đối với các thuốc này và do đó không xuất hiện trong bối cảnh này.
Trong các thuốc ức chế EGFR hiện có, sự kết hợp giữa EGFR TKI thế hệ một và thế hệ ba đã cho thấy một số hoạt tính ngắn hạn ở bệnh nhân mang đột biến trans T790M và C797S. Tuy nhiên, cách tiếp cận này không hiệu quả ở bệnh nhân có đột biến cis T790M và C797S, trong khi brigatinib kết hợp cetuximab là một lựa chọn tiềm năng cho nhóm này. Trong một nghiên cứu hồi cứu trên 15 bệnh nhân ung thư phổi biểu mô tuyến giai đoạn tiến xa mang đột biến EGFR C797S sau điều trị bằng osimertinib,năm bệnh nhân được điều trị bằng brigatinib cộng cetuximab, trong đó ba bệnh nhân đạt đáp ứng một phần (PR) và hai bệnh nhân đạt bệnh ổn định (SD). Ngược lại, ở các quần thể không chọn lọc, các liệu pháp kết hợp osimertinib với EGFRTKIs thế hệ hai cho thấy hiệu quả hạn chế và lo ngại về an toàn. Ví dụ, trong một thử nghiệm pha I trên 13 bệnh nhân NSCLC dương tính EGFR kháng osimertinib,việc kết hợp afatinib với osimertinib chỉ đạt tỷ lệ đáp ứng khách quan (ORR)7,7% và tỷ lệ kiểm soát bệnh (DCR) 46,2%; các tác dụng phụ bao gồm tiêu chảy ở 76,9% bệnh nhân (độ 3–4 ở 15,4%), thiếu máu ở 76,9% (tất cả ≤ độ 2) và phát ban ở 69,2% (độ 3–4 ở 7,7%).
Kháng thể hai đặc hiệu EGFR–MET: Liệu pháp nhắm kép EGFR và MET bằng amivantamab đã chứng minh hiệu quả trong điều trị NSCLC kháng EGFR TKIs. Trong thử nghiệm pha I CHRYSALIS, amivantamab đơntrị đạt ORR 19% và thời gian đáp ứng (DOR) trung vị 5,9 tháng ở 121 bệnh nhân tiến triển sau osimertinib. Trong số 45 bệnh nhân được điều trị bằng amivantamab cộng lazertinib, ORR đạt 36% với DOR trung vị 9,6 tháng, cho thấy tiềm năng của liệu pháp kết hợp.
Thử nghiệm pha III MARIPOSA-2 đã phân ngẫu nhiên 657 bệnh nhân NSCLC giai đoạn tiến xa đã điều trị bằng osimertinib (tỷ lệ 2:2:1) vào ba nhóm: amivantamab + lazertinib + hóa trị, hóa trị đơn thuần hoặc amivantamab + hóa trị. Kết quả cho thấy PFS trung vị là 8,3 tháng với phác đồ amivantamab–lazertinib–hóa trị (HR 0,44; P < 0,001) và 6,3 tháng với amivantamab–hóa trị (HR 0,48; P < 0,001), so với 4,2 tháng với hóa trị đơn thuần, chứng minh hiệu quả vượt trội của các phác đồ chứa amivantamab.Tuy nhiên, tỷ lệ tác dụng phụ xuất hiện trong quá trình điều trị (TEAEs) độ ≥3cao hơn đáng kể ở các nhóm chứa amivantamab (92% và 72% so với 48% ở nhóm hóa trị đơn thuần). FDA đã phê duyệt phác đồ amivantamab–hóa trị (không kèm lazertinib) cho điều trị hàng hai NSCLC giai đoạn tiến xa mang đột biến EGFR.
Đối với bệnh nhân kháng osimertinib sau điều trị đầu tay, hóa trị xen kẽ có thể loại bỏ các dòng kháng thuốc, tiềm năng giúp khối u nhạy trở lại với ức chế EGFR. Tuy nhiên, việc tái điều trị bằng osimertinib đơn độc dường như không phải là lựa chọn khả thi. Một nghiên cứu hồi cứu tại một trung tâm trên 17 bệnh nhân được tái điều trị bằng osimertinib cho thấy chỉ có 2 bệnh nhân (13%) đạt PR, 6 bệnh nhân (38%) đạt SD, và thời gian trung vị của liệu pháp tái điều trị chỉ là 3,6 tháng.
Vượt qua kháng thuốc ngoài đích off-target – Các biến đổi MET Khuếch đại và/hoặc tăng biểu hiện MET là một cơ chế quan trọng gây kháng mắc phải đối với EGFRTKIs. Nhiều nghiên cứu lâm sàng đã khảo sát các chiến lược kết hợp MET TKIs và EGFR TKIs nhằm xử lý cơ chế kháng này. Đáng chú ý, các nghiên cứu khác nhau đã sử dụng định nghĩa khác nhau về khuếch đại và/hoặc tăng biểu hiện MET.
Trong các thử nghiệm TATTON (pha Ib) và SAVANNAH (pha II), thuốc MET TKI savolitinib kết hợp với osimertinib được đánh giá ở bệnh nhân NSCLC mang đột biến EGFR và khuếch đại/tăng biểu hiện MET sau khi bệnh tiến triển trên EGFR TKI trước đó.
● Trong TATTON, khuếch đại MET được định nghĩa bằng FISH (Fluorescence In Situ Hybridization - Lai huỳnh quang tại chỗ) là số bản sao MET ≥5 hoặc tỷ lệMET:CEP7 ≥2, hoặc bằng NGS là ≥5 bản sao MET so với ploidy khối u (với độ sâu giải trình tự ≥200× trong mẫu có ≥20% tế bào u), và tăng biểu hiện MET bằng IHC3+ trên ≥50% tế bào u (ISH - In Situ Hybridization - lai tại chỗ). Kết quả: ORR 33% ở bệnh nhân đã dùng EGFR TKI thế hệ 3 và 62–67% ở bệnh nhân chưa dùng, vớiPFS trung vị 5,5 tháng và 9,0–11,1 tháng.
● Trong SAVANNAH, tiêu chuẩn IHC 90+ (IHC 3+ trên ≥90% tế bào u) hoặc FISH 10+ (số bản sao MET ≥10) được xác định là tiêu chí dự đoán đáp ứng tốt nhất. Trong nhóm hiệu quả chính gồm 80 bệnh nhân, ORR đạt 56%, DOR trung vị 7,1 tháng và PFS trung vị 7,4 tháng. Các tác dụng phụ thường gặp: phù ngoại vi (46%), buồn nôn(40,5%), tiêu chảy (23,2%). Đáng chú ý, ở 25 bệnh nhân IHC 90+ hoặc FISH 10+dùng savolitinib đơn trị, ORR chỉ đạt 16%, PFS trung vị 3,6 tháng, nhấn mạnh tầm quan trọng của việc duy trì ức chế EGFR.
INSIGHT2 (pha II) đánh giá tepotinib + osimertinib sau tiến triển trên osimertinib đầu tay ở 98 bệnh nhân có khuếch đại MET (FISH ≥5 bản sao hoặc MET:CEP7 ≥2, hoặc NGS sinh thiết lỏng ≥2,3 bản sao MET). Kết quả: ORR 50%, PFS trung vị 5,6 tháng. Các thuốc MET khác như capmatinib, vebreltinib (bozitinib), gumarontinib (glumetinib) cũng cho thấy hiệu quả hứa hẹn khi kết hợp EGFR TKIs,với ORR dao động 40–60%. Tuy nhiên, hầu hết các nghiên cứu đều là thử nghiệm đơn nhánh, không ngẫu nhiên, cỡ mẫu nhỏ.
SACHI (pha III) là thử nghiệm đầu tiên so sánh osimertinib + savolitinib với hóa trị chuẩn line-2 ở bệnh nhân có khuếch đại MET sau tiến triển trên EGFR TKI. Kết quả: Ở bệnh nhân đã dùng EGFR TKI thế hệ 3: PFS trung vị 6,9 tháng so với 3,0 tháng (HR 0,32; P < 0,0001). Ở bệnh nhân đã dùng EGFR TKI thế hệ trước: PFS trung vị 8,2 tháng so với 5,7 tháng (HR 0,47; P =0,0017). ORR cũng cao hơn rõ rệt với phác đồ kết hợp. Xu hướng cải thiện OS cũng được ghi nhận. Sau SACHI, chiến lược này đang được đánh giá trong thử nghiệm toàn cầu SAFFRON (pha III).
Telisotuzumab vedotin (Teliso-V) là một ADC gồm kháng thể kháng MET, linker có thể cắt và payload gây độc tế bào MMAE. Kết hợp Teliso-V + osimertinib ở 38 bệnh nhân NSCLC đột biến EGFR và tăng biểu hiện MET sau osimertinib cho ORR 50%, PFS trung vị 7,4 tháng. Tác dụng phụ: bệnh lý thần kinh ngoại vi (50%),phù ngoại vi (32%), viêm phổi mức độ thấp (5%). Telisotuzumab adizutecan (Temab-A) là ADC khác dựa trên cùng kháng thể nhưng payload là chất ức chế TOPO1. Trong thử nghiệm pha I trên 41 bệnh nhân NSCLC đột biến EGFR sau EGFR TKI và hóa trị, ORR đạt 63%, PFS trung vị 10,9 tháng, hiệu quả cao bất kể mức biểu hiện MET. Tác dụng phụ chủ yếu về huyết học và tiêu hóa; ILD/viêm phổi xảy ra ở 7% (1 ca tử vong).
Cơ chế kháng EGFR–MET kép. Phân tích ctDNA từ INSIGHT2 cho thấy trong 29 bệnh nhân tiến triển trên tepotinib + osimertinib: 34% có đột biến kháng on-target ở EGFR và/hoặc MET, 31% có đột biến EGFR (5 ca C797S, 3 ca đã có từ trước), 14%có đột biến mới tại miền kinase MET (D1228N/H/Y, Y1230H), 38% có đột biến kích hoạt đường tín hiệu bypass (KRAS, MYC, BRAF, PIK3CA); mất khuếch đại MET ở 9/12 bệnh nhân, trong khi 3 bệnh nhân lại có tăng khuếch đại MET.
Chiến lược sau thất bại EGFR–MET kép. Hiện chưa có nhiều nghiên cứu tiền cứu. Một phân tích hồi cứu trên 77 bệnh nhân NSCLC đột biến EGFR và mắc phải khuếch đại MET cho thấy: sau thất bại EGFR–MET kép, điều trị cứu vãn bằng TKI hoặc hóa trị cải thiện PFS sau tiến triển (3,9 và 4,9 tháng) so với chăm sóc hỗ trợ (1,5 tháng); OS sau tiến triển cũng cao hơn (9,2 và 7,7 tháng so với 2,3 tháng). Tuy nhiên, cần thêm nghiên cứu để xác định chiến lược tối ưu cho bệnh nhân sau thất bại liệu pháp kép EGFR–MET.
Liệu pháp nhắm RTK fusion. Các RTK fusion, bao gồm ALK, RET hoặc FGFR3, là một cơ chế quan trọng gây kháng bypass đối với EGFR TKIs. Các nghiên cứu ban đầu, chủ yếu là báo cáo ca bệnh, cho thấy liệu pháp TKI kép nhắm đồng thời RTK fusion mắc phải và oncogenic driver ban đầu có thể mang lại lợi ích lâm sàng.Trong một tổng quan y văn, kết quả từ 50 bệnh nhân có RTK fusion mắc phải sau điều trị EGFR TKI cho thấy: 1 bệnh nhân (2%) đạt đáp ứng hoàn toàn (CR), 29(58%) đáp ứng một phần (PR), 13 (26%) bệnh ổn định (SD) và 7 (14%) bệnh tiến triển; thời gian đáp ứng (DOR) dài nhất đạt 25 tháng. Tương tự, Zhu và cộng sự báo cáo trên 86 bệnh nhân có RTK fusion liên quan đến kháng thứ phát EGFR TKIs, trong đó 10 bệnh nhân được điều trị TKI kép: 6 (60%) đạt PR, 4 (40%) đạt SD, DOR dài nhất 12 tháng. Một nghiên cứu tiền cứu trên 14 bệnh nhân mang đột biến EGFR và RET fusion sau tiến triển trên osimertinib cho thấy kết hợp osimertinib+ selpercatinib đạt ORR 50%, DCR 83% và thời gian điều trị trung vị 7,9 tháng,không ghi nhận tác dụng phụ nghiêm trọng liên quan điều trị. Tuy nhiên, trong một phân tích hồi cứu trên 27 bệnh nhân có RTK fusion mắc phải, 14 bệnh nhân dùng TKI kép chỉ đạt ORR 21,4% và DCR 78,6% — thấp hơn các báo cáo ca bệnh khác. Sự khác biệt này có thể do thiên lệch công bố (ưu tiên báo cáo kết quả tích cực), sự khác nhau về loại RTK fusion và yếu tố bệnh học. Nhìn chung, dữ liệu cho thấy ức chế kép EGFR–RTK là khả thi và hiệu quả ở bệnh nhân NSCLC có RTK fusion mắc phải sau EGFR TKI, với CR hiếm nhưng PR và SD phổ biến, một số bệnh nhân có đáp ứng dài hạn.
Các biến đổi HER2: khuếch đại HER2 được phát hiện ở khoảng 2% bệnh nhân NSCLC đột biến EGFR kháng osimertinib line-1 và 5% sau osimertinib line-2. Đột biến HER2, chủ yếu là exon 20 insertion, chiếm 1,5% tổng số NSCLC. Ngoài ra, HER2 mutation mắc phải được báo cáo ở 1% bệnh nhân đột biến EGFR sau osimertinib line-1. Tăng bộc lộ HER2 được quan sát ở khoảng 13% bệnh nhân tiến triển trên EGFR TKIs. Nhiều ADC và TKI nhắm HER2 (như zongertinib) đã chứng minh hiệu quả ở bệnh nhân NSCLC đột biến HER2 de novo. Thử nghiệm TRAEMOS (phaI/II) đánh giá ADC trastuzumab emtansine (T-DM1) kết hợp osimertinib ở 27 bệnh nhân NSCLC EGFR-mutant, HER2 overexpression sau tiến triển trên osimertinib. Hiệu quả hạn chế: ORR 4% sau 12 tuần, DCR 33%, PFS trung vị 2,8 tháng. DESTINY-Lung01 (pha II) đánh giá trastuzumab deruxtecan (T-DXd) đơn trị ở 41 bệnh nhân NSCLC HER2-overexpression, ORR 34,1% (2% CR), DCR 78%, PFS trung vị 6,7 tháng, OS trung vị 11,2 tháng. DESTINY-Lung03 (pha Ib) trên 19 bệnh nhân bộc lộ quá mức HER2 sau EGFR TKI cho thấy T-DXd đạt ORR 68,4%, DCR 84,2%, PFS trung vị 8,2 tháng, OS trung vị 19,6 tháng. Dù cỡ mẫu nhỏ, dữ liệu cho thấy T-DXd có hiệu quả hứa hẹn ở bệnh nhân bộc lộ quá mức HER2 sau EGFR TKI. FDA đã phê duyệt T-DXd theo hướng không phụ thuộc mô bệnh học cho bệnh nhân ung thư giai đoạn tiến xa HER2 IHC 3+. Thử nghiệm DESTINY-PanTumor03 (pha II) đang tiến hành tại Trung Quốc để đánh giá T-DXd ± bevacizumab ở bệnh nhân ung thư có bộc lộ HER2, bao gồm NSCLC.
Chiến lược không dựa trên biomarker để vượt qua kháng EGFR TKI – ADCs.
● TROP2-targeted ADCs. ADC datopotamab deruxtecan (Dato-DXd) được đánh giá trong TROPION-Lung01 (pha III), thử nghiệm đầu tiên về ADC nhắm TROP2 ở NSCLC. Trong 98 bệnh nhân đã điều trị, 84 có đột biến EGFR, PFS trung vị 5,7 tháng với Dato-DXd so với 2,6 tháng với docetaxel. Phân tích gộp trên 117 bệnh nhân đột biến EGFR NSCLC (96 tiến triển sau osimertinib) cho thấy ORR 43%, DCR 86%, DOR trung vị 7 tháng, PFS 5,8 tháng, OS 15,6 tháng. Dựa trên dữ liệu này, FDA đã phê duyệt Dato-DXd cho bệnh nhân NSCLC đột biến EGFR giai đoạn tiến xa sau EGFR TKI và hóa trị platinum. Trong ORCHARD (pha II), kết hợp osimertinib + Dato-DXd đạt ORR 36%, PFS trung vị 11,7 tháng.Các thử nghiệm TROPION-Lung14/15 (pha III) đang tiếp tục đánh giá Dato-DXd ± osimertinib ở bệnh nhân đột biến EGFR NSCLC. Tại Trung Quốc, ADC sacituzumab tirumotecan (sac-TMT) đã được phê duyệt cho NSCLC đột biến EGFR tiến triển sau EGFR TKI. Trong OptiTROP-Lung04 (pha III), sac-TMT cải thiện PFS (8,3 vs 4,3 tháng) và OS (NR vs 17,4 tháng). Các thử nghiệm đang tiếp tục so sánh sac-TMTvới docetaxel/pemetrexed hoặc kết hợp với carboplatin, hoặc với kháng thể PD-L1 tagitanlimab.
● Đích HER3 ADCs. HER3, một thành viên khác của họ HER, đóng vai trò quan trọng trong NSCLC EGFR-mutant, thúc đẩy tín hiệu PI3K–AKT, cơ chế kháng EGFR TKIs. HER3 expression tăng sau kháng EGFRTKIs, khiến HER3 trở thành đích tiềm năng. ADC patritumab deruxtecan (HER3-DXd)đã cho thấy hoạt tính lâm sàng đáng khích lệ. Trong HERTHENA-Lung01 (pha II)trên 225 bệnh nhân NSCLC đột biến EGFR tiến triển sau EGFR TKIs và hóa trị platinum (209 tiến triển trên osimertinib), ORR đạt 29,2%, DCR 72,7%, PFS trung vị 5,5 tháng, OS trung vị 11,9 tháng. TEAEs độ ≥3 xảy ra ở 64,9% bệnh nhân, chủ yếu độc tính huyết học; ILD xảy ra ở 5,3% (1 ca tử vong). Phân tích biomarkercho thấy đáp ứng với HER3-DXd xảy ra bất kể mức biểu hiện HER3. HERTHENA-Lung02 (pha III) so sánh HER3-DXd với hóa trị pemetrexed–platinum ở 586 bệnh nhân NSCLC đột biến EGFR sau tiến triển trên EGFR TKI thế hệ 3. Kết quả: ORR 35,2% vs 25,3%, PFS 5,8 vs 5,4 tháng, nhưng chưa có lợi ích OS. Do đó, nhà tài trợ đã rút hồ sơ xin FDA phê duyệt. Tuy nhiên, các phân tích biomarker đang tiếp tục để tinh chỉnh chọn bệnh nhân.
● Các ADC khác nhắm HER3. Các ADC nhắm HER3 khác, như SHR-A2009 (NCT06465238) và YL202/BNT326 (NCT07111520), đang được nghiên cứu trong các thử nghiệm pha I ở bệnh nhân NSCLC kháng EGFR TKIs. SHR-A2009 có payload là chất ức chế TOPO1.Trong một thử nghiệm pha I trên bệnh nhân ung thư giai đoạn tiến xa, có 36 bệnh nhân NSCLC, trong đó 34 bệnh nhân mang đột biến EGFR đã được điều trị bằng EGFRTKIs, bao gồm 29 bệnh nhân kháng EGFR TKI thế hệ thứ ba. Trong số 30 bệnh nhân có thể đánh giá, ORR đạt 30,0%, DCR 76,7%, DOR trung vị 7,0 tháng, và tỷ lệ PFS6 tháng là 49,8%. Tương tự HER3-DXd, độc tính huyết học là tác dụng phụ thường gặp, và ILD xảy ra ở 4,8% bệnh nhân.
● ADC nhắm kép EGFR–HER3. Các ADC nhắm kép EGFR–HER3 đang được phát triển lâm sàng. Izalontamab brengitecan (iza-bren, trước đây là BL-B01D1), ADC nhắm kép EGFR–HER3 đầu tiên với payload là chất ức chế TOPO1, đã được đánh giá trong thử nghiệm pha I trên 195 bệnh nhân ung thư giai đoạn tiến xa kháng trị. Trong nhóm gồm 41 bệnh nhân NSCLC đột biến EGFR (37 bệnh nhân từng điều trị bằng EGFR TKI thế hệ ba), ORR đạt 67,5%, DCR 87,5%, PFS trung vị 5,6 tháng, DOR trung vị 8,5 tháng, cho thấy tiềm năng điều trị đáng hứa hẹn cho nhóm bệnh nhân kháng EGFRTKIs.
● Ngoài ra, thử nghiệm pha Ib/II đánh giá SYS6010, một ADC mới nhắm EGFR với payload TOPO1, cũng cho thấy hiệu quả khả quan ở bệnh nhân NSCLC giai đoạn tiến xa mang đột biến EGFR. ORR và DCR lần lượt đạt 90% và 100% ở 10 bệnh nhân chỉ từng điều trị bằng EGFR TKIs, và 41,5% và 90,2% ở 41 bệnh nhân đã từng điều trị bằng EGFR TKIs và hóa trị platinum. Các thử nghiệm lâm sàng tiếp theo đang được tiến hành để xác nhận giá trị điều trị của các ADC nhắm EGFR.
Thuốc kháng tạo mạch (Anti-angiogenic agents). Các thụ thể EGFR và VEGF (VEGFRs) có chung nhiều con đường tín hiệu hạ lưu. Các nghiên cứu tiền lâm sàng đã chứng minh rằng biểu hiện VEGF tăng đáng kể trong tế bào NSCLC sau khi xuất hiện kháng EGFR TKIs, tạo cơ sở hợp lý cho các liệu pháp kết hợp nhắm vào trục EGFR–VEGF. Bằng chứng tiền lâm sàng cho thấy TKI nhắm VEGFR anlotinib có hoạt tính chống u hiệp đồng khi kết hợp với osimertinib. Một nghiên cứu hồi cứu đa trung tâm trên 268 bệnh nhân NSCLC đột biến EGFR kháng osimertinib cho thấy PFS trung vị ởnhóm dùng anlotinib đơn trị hoặc anlotinib + osimertinib đạt 6,9 tháng. Tương tự, một nghiên cứu hồi cứu khác trên 39 bệnh nhân NSCLC đột biến EGFR kháng osimertinib, điều trị bằng apatinib + osimertinib, ghi nhận ORR 12,8%, DCR79,5% và PFS trung vị 4 tháng. Tuy nhiên, các kết quả này cần được xác nhận trong các thử nghiệm tiền cứu quy mô lớn.
Thuốc ức chế điểm kiểm soát miễn dịch (Immune-checkpoint inhibitors – ICIs): Đơn trị ICI có hiệu quả hạn chế ở bệnh nhân NSCLC đột biến EGFR kháng EGFR TKIs, do đó nhóm bệnh nhân này thường bị loại trừ khỏi điều trị ICI. Các chiến lược ICI kép cũng không mang lại lợi ích sống còn. Thử nghiệm pha III CheckMate 722 và KEYNOTE-789 cho thấy việc thêm kháng thể kháng PD-1 (nivolumab hoặc pembrolizumab) vào hóa trị không cải thiện PFS hay OS.
Các chiến lược kết hợp.
● Phân tích thăm dò từ thử nghiệm pha III IMpower150 cho thấy kết hợp kháng thể kháng PD-L1 atezolizumab với bevacizumab + carboplatin–paclitaxel (ABCP) cải thiện PFS (9,7vs 6,1 tháng; HR 0,42) nhưng không cải thiện OS (27,8 vs 18,1 tháng).
● Thử nghiệm pha III ATTLAS và ORIENT-31 chứng minh rằng thêm kháng thể kháng PD-(L)1 và bevacizumab (hoặc thuốc tương tự) vào hóa trị giúp cải thiện ORR và PFS so với hóa trị đơn thuần ở bệnh nhân NSCLC đột biến EGFR kháng EGFR TKIs.
● Ngược lại, thử nghiệm IMpower151 (tại Trung Quốc) cho thấy ABCP không cải thiện PFS so với BCP ở bệnh nhân NSCLC đột biến EGFR hoặc tái sắp xếp ALK.
● Một phân tích gộp từ 17 nghiên cứu đơn nhánh và 15 thử nghiệm ngẫu nhiên (tổng cộng 2.886 bệnh nhân) cho thấy kết hợp ICI + thuốc kháng tạo mạch + hóa trị vượt trội hơn so với ICI + hóa trị, các phác đồ không ICI hoặc ICI đơn trị về ORR,DCR và PFS. Tuy nhiên, chiến lược này cũng đi kèm với tăng tỷ lệ tác dụng phụ (TRAEs), ngừng điều trị và điều chỉnh liều.
Ivonescimab. Đây là kháng thể kép người hóa nhắm đồng thời PD-1 và VEGFA. Trong thử nghiệm pha III HARMONi-A, ivonescimab + hóa trị cải thiện PFS so với hóa trị đơn thuần ở bệnh nhân NSCLC đột biến EGFR kháng EGFR TKIs (7,1 vs 4,8 tháng; HR 0,46; P < 0,0001). Lợi ích tương tự được ghi nhận ở bệnh nhân mang đột biến 19del, di căn não, đã điều trị bằng EGFR TKI thế hệ 3 và có đồng thời T790M. Dữ liệu OS còn chưa chín muồi nhưng có xu hướng ủng hộ nhóm ivonescimab. Đáng chú ý, ở bệnh nhân EGFR T790M dương tính, ivonescimab cải thiện PFS rõ rệt (HR 0,22), trong khi lợi ích của sintilimab + bevacizumab biosimilar IBI305 trongORIENT-31 rất hạn chế (HR 1,10). Thử nghiệm mở rộng toàn cầu HARMONi (NCT06396065) đang được tiến hành.
Các thuốc ức chế điểm kiểm soát miễn dịch (ICIs) dạng kháng thể kép khác. SHR-1701, một tác nhân kháng thể kép mới kết hợp kháng thể đơn dòng kháng PD-L1 với miền ngoại bào của thụ thể TGFβ II, đã cho thấy ORR 16,7% và DCR 50,0% khi dùng đơn trị ở 24 bệnh nhân NSCLC kháng EGFR TKIs trong một thử nghiệm pha I đa trung tâm. KN046, một kháng thể kép PD-L1–CTLA4, đã được thử nghiệm trong một nghiên cứu pha II trên bệnh nhân NSCLC đột biến EGFR đã điều trị bằng EGFR TKIs (nhưng chưa hóa trị). Cụ thể, 26 bệnh nhân được điều trị bằng KN046 kết hợp hóa trị nền platinum, kết quả đạt ORR 26,9%, DCR 84,6%, PFS trung vị 5,5 tháng và OS trung vị 20,2 tháng.Nhìn chung, các ICI kháng thể kép cho thấy nhiều hứa hẹn trong việc tái cấu trúc vi môi trường khối u (TME) ức chế miễn dịch ở NSCLC đột biến EGFR, có thể mang lại lợi ích sống còn vượt trội so với các ICI truyền thống. Tuy nhiên,những kết quả ban đầu này cần được xác nhận trong các thử nghiệm lâm sàng quy mô lớn. Hiện tại, một thử nghiệm pha II đang tiến hành đánh giá cadonilimab (kháng thể kép PD-L1–CTLA4) kết hợp anlotinib và pemetrexed ở bệnh nhân NSCLC kháng EGFR TKIs, âm tính với EGFRT790M (NCT06277674).
4. Chiến lược đối phó với oligoprogression và tiến triển tại hệ thần kinh trung ương (CNS)
Oligoprogression: là tiến triển trên hình ảnh học và/hoặc xuất hiện mới≤5 tổn thương di căn trong hoặc sau điều trị toàn thân, sau khi đã đạt được đáp ứng hoàn toàn (CR) hoặc một phần (PR). Oligoprogression khá phổ biến sau đơntrị liệu bằng osimertinib, xảy ra ở khoảng 49–73% bệnh nhân. Các phương pháp điều trị tại chỗ như xạ trị định vị thân (SBRT) hoặc ít gặp hơn là phẫu thuật cắt bỏ được khuyến nghị cho nhóm bệnh nhân này, kết hợp với việc tiếp tục dùng osimertinib. Lý do là liệu pháp tại chỗ có thể loại bỏ các dòng tế bào kháng thuốc tại vị trí khối u tiến triển, trong khi các vị trí khác vẫn nhạy cảm với osimertinib. Nhiều nghiên cứu cho thấy liệu pháp tại chỗ kết hợp tiếp tục TKI có thể đạt PFS lần hai từ 6–10 tháng, giúp trì hoãn nhu cầu chuyển sang hóa trị. Thử nghiệm pha II/III HALT (NCT03256981) đang ngẫu nhiên phân nhóm bệnh nhân NSCLC do oncogene với oligoprogression để tiếp tục TKI có hoặc không kèm SBRT.
Tiến triển tại CNS (Central Nerves System – hệ thần kinh trung ương): Mặc dù EGFR TKIs thế hệ ba có khả năng thấm qua hàng rào máu não và hoạt tính nộisọ đáng kể, khoảng 5–30% bệnh nhân điều trị bằng osimertinib đầu tay vẫn tiến triển tại CNS. Cơ chế kháng này chưa được hiểu rõ do việc sinh thiết lại di căn não còn hạn chế. Xạ phẫu định vị và xạ trị toàn não vẫn là tiêu chuẩn cho di căn não khu trú hoặc lan tỏa. Về điều trị toàn thân, tiếp tục osimertinib sau tiến triển di căn não kết hợp hóa trị dường như cải thiện kiểm soát bệnh nội sọ, và nhiều bác sĩ lâm sàng áp dụng cách này. Ở bệnh nhân có khuếch đại MET và di căn não sau osimertinib đầu tay, phác đồ osimertinib + savolitinib second-line đạt ORR nội sọ 43% (6/14 bệnh nhân) trong thử nghiệm SAVANNAH. Ngoài ra, các ADC cũng cho thấy hoạt tính CNS: trong HERTHENA-Lung01, HER3-DXd đạt ORR nội sọ 33% ở nhóm 30 bệnh nhân có di căn não chưa xạ trị; trong TROPION-Lung05, Dato-DXd đạt ORR nội sọ 22% ở 18 bệnh nhân. Với kháng thể kép, amivantamab +hóa trị ± lazertinib kéo dài PFS nội sọ trong thử nghiệm MARIPOSA-2 (12,8 và 12,5 tháng so với 8,3 tháng ở nhóm hóa trị đơn thuần), mặc dù tỷ lệ đáp ứng nội sọ chưa được báo cáo.
Ngoài di căn nhu mô não, di căn màng não (LMD –leptomeningeal disease) là biến chứng nặng nề hơn sau kháng EGFR TKIs. Một phân tích hồi cứu trên nhóm lớn cho thấy tiếp tục EGFR TKIs thế hệ ba qua được hàng rào máu não sau LMD cải thiện OS đáng kể so với nhóm không tiếp tục (23,2vs 8,8 tháng ở nhóm chưa dùng TKI qua được hàng rào máu não trước đó; 12,4 vs 6 tháng ở nhóm đã dùng trước đó; P < 0,001). Tuy nhiên, chiến lược này chỉ trì hoãn chứ không ngăn ngừa được sự xuất hiện di căn màng não.
Chiến lược trì hoãn kháng EGFR TKIs. Ngoài việc xử lý kháng thuốc sau khi xảy ra,trì hoãn kháng thuốc là một chiến lược quan trọng nhằm cải thiện kết quả cho bệnh nhân. Trì hoãn kháng thuốc tập trung vào việc chủ động triển khai các liệu pháp kết hợp để kéo dài thời gian đáp ứng. Các cách tiếp cận hiện nay gồm: Ngăn chặn tiến hóa dòng tế bào và loại bỏ DTPs (drug-tolerant persisters), chặn trước các con đường kháng thuốc, phát triển EGFR TKIs mạnh hơn để ức chế khángon-target phụ thuộc EGFR. Do chưa có EGFR TKIs nào mạnh hơn thế hệ ba được đưa vào thực hành lâm sàng, thảo luận tập trung vào hai chiến lược đầu tiên.
Ngăn chặn tiến hóa dòng tế bào và loại bỏ DTPs. DTPs ngày càng được công nhận là yếu tố trung gian quan trọng của kháng thuốc và tái phát. Đây là quần thể tế bào có khả năng tồn tại dưới áp lực điều trị, đặc trưng bởi sự tăng sinh chậm và thay đổi kiểu hình có thể đảo ngược, thay vì đột biến gen mắc phải không thể đảo ngược. Cơ chế DTPs dẫn đến kháng thuốc thật sự rất đa dạng: ngủ đông, tái cấu trúc biểu sinh, EMT, thay đổi chuyển hóa, tự thực, kháng apoptosis. Chiến lược hiện tại gồm: loại bỏ chọn lọc DTPs bằng cách nhắm vào các con đường đặc trưng, loại bỏ tổng quát DTPs bằng các liệu pháp gây độc tế bào rộng.
Các nghiên cứu tiền lâm sàng cho thấy thêm CDK4/6 inhibitors vào osimertinib có thể phá vỡ trạng thái ngủ đông của DTPs, ức chế chu kỳ tế bào và thúc đẩy apoptosis; kết hợp EGFR TKIs với KDM5A inhibitors hoặc HDAC inhibitors cũng loại bỏ DTPs, ức chế các con đường EMT và liên quan tế bào gốc (AXL, FGFR, YAP–TEAD, Wnt–β-catenin) có tiềm năng loại bỏ DTPs và giảm kháng EGFR TKIs thế hệ ba, đối kháng HGF và IGF1 (ligands của MET và IGF1R) do fibroblast liên quan ung thư tiết ra có thể tái nhạy cảm tế bào NSCLC đột biến EGFR với EGFR TKIs bằng cách ức chế EMT qua trung gian cận tiết và kiểu hình kháng thuốc liên quan.
5. Từ khái niệm tiến triển trên hình ảnh đến theo dõi động học kháng thuốc ở mức độ phân tử
5.1. Hạn chế của mô hình theo dõi tiến triển truyền thống
Trong thực hành lâm sàng hiện nay, đánh giá tiến triển bệnh ở bệnh nhân ung thư phổi không tế bào nhỏ (NSCLC) chủ yếu dựa trên hình ảnh học theo tiêu chuẩn RECIST 1.1, kết hợp với triệu chứng lâm sàng và một số chỉ điểm sinh học không đặc hiệu. Mặc dù mô hình này đơn giản và dễ áp dụng, nó tồn tại nhiều hạn chế căn bản trong bối cảnh điều trị đích hiện đại, đặc biệt ở nhóm NSCLC đột biến EGFR. Thứ nhất, mô hình theo dõi truyền thống chịu ảnh hưởng rõ rệt của độ trễ sinh học. Các nghiên cứu cho thấy kháng thuốc ở mức phân tử thường xuất hiện từ hàng tuần đến hàng tháng trước khi có bất kỳ thay đổi nào được ghi nhận trên CT hoặc MRI. Nói cách khác, kháng thuốc phân tử luôn đi trước tiến triển hình ảnh, khiến việc chờ đợi RECIST tiến triển có thể làm mất đi “cửa sổ can thiệp” quan trọng. Thứ hai, đánh giá dựa trên hình ảnh không phản ánh đầy đủ dị hợp tính khối u (tumor heterogeneity). Trong cùng một thời điểm, có thể tồn tại tình trạng một số tổn thương tiến triển trong khi các tổn thương khác vẫn ổn định. Đồng thời, các clone kháng thuốc có thể tồn tại ở mức rất thấp, không đủ tạo ra thay đổi kích thước khối u nhưng vẫn có khả năng chi phối diễn tiến bệnh về sau. Những clone “nhỏ nhưng nguy hiểm” này hầu như không thể được phát hiện bằng phương pháp hình ảnh học đơn thuần. Cuối cùng, RECIST chỉ trả lời câu hỏi mang tính mô tả là “khối u có tolên hay không”, mà không cung cấp thông tin mang tính cơ chế như “tại sao khối u kháng thuốc”. Điều này hạn chế khả năng định hướng chiến lược điều trị tiếp theo trong kỷ nguyên điều trị cá thể hóa.
5.2. Khái niệm mới: Molecular progression
Từ những hạn chế trên, khái niệm molecular progression đã được đề xuất nhằm tái định nghĩa tiến triển bệnh trong ung thư học hiện đại. Molecular progression được hiểu là sự xuất hiện hoặc gia tăng của các dòng kháng thuốc ở mức phân tử, ngay cả khi bệnh nhân chưa có biểu hiện lâm sàng hoặc tiến triển trên hình ảnh học. Khái niệm này dẫn đến một mô hình phân tầng tiến triển mới, trong đó tiến triển bệnh được xem xét ở ba cấp độ:cấp độ phân tử (dựa trên ctDNA và các đột biến gen), cấp độ hình ảnh (CT, MRI, PET) và cấp độ lâm sàng (triệu chứng). Trong mô hình này, tiến triển phân tử được xem là giai đoạn sớm nhất và có giá trị dự báo cao nhất cho diễn tiến bệnh về sau.
Liquid biopsy (sinh thiết lỏng) và ctDNA – nền tảng của theo dõi động học kháng thuốc
Liquid biopsy, đặc biệt là phân tích circulating tumor DNA (ctDNA), đóng vai trò trung tâm trong việc phát hiện tiến triển phân tử. ctDNA là các mảnh DNA có nguồn gốc từ tế bào ung thư, lưu hành trong máu ngoại vi, được giải phóng thông qua các quá trình apoptosis, hoại tử (necrosis) hoặc bài tiết chủ động của tế bào u.
Ưu điểm nổi bật của ctDNA là tính không xâm lấn, cho phép lấy mẫu lặp lại nhiều lần trong quá trình điều trị. Ngoài ra, ctDNA phản ánh tương đối toàn diện gánh nặng khối u của toàn cơ thể, không phụ thuộc vào một vị trí sinh thiết đơn lẻ. Quan trọng hơn, ctDNA có khả năng phát hiện sớm các đột biến kháng thuốc trước khi xuất hiện tiến triển trên hình ảnh, đồng thời cho phép theo dõi động học tiến hóa của khối u theo thời gian thực.
Trong NSCLC đột biến EGFR, phân tích ctDNA có thể phát hiện các đột biến kháng thuốc điển hình như T790M hoặc C797S, các cơ chế bypass như khuếch đại MET hay đột biến KRAS, đánh giá tình trạng bệnh tồn dư tối thiểu (MRD) sau điều trị triệt căn, cũng như theo dõi đáp ứng sớm với TKI.
Minimal Residual Disease (MRD) – khái niệm mới trong ung thư phổi
Minimal residual disease (MRD – bệnh tồn dư tối thiểu) được định nghĩa là sự tồn tại của các tế bào ung thư còn sót lại sau điều trị triệt căn, ở mức độ không thể phát hiện bằng các phương tiện hình ảnh thông thường. Trong những năm gần đây, MRD ngày càng được công nhận là một yếu tố tiên lượng quan trọng trong ung thư phổi. Nhiều nghiên cứu cho thấy bệnh nhân có MRD dương tính, được xác định thông qua ctDNA, có nguy cơ tái phát cao hơn rõ rệt so với nhóm MRD âm tính, đồng thời có thời gian sống không bệnh ngắn hơn. Trong bối cảnh NSCLC đột biến EGFR sau phẫu thuật ± điều trị bổ trợ, việc phát hiện ctDNA dương tính có thể gợi ý nhu cầu điều trị bổ trợ tích cực hơn,trong khi ctDNA âm tính cho phép chiến lược theo dõi chặt chẽ. Cách tiếp cận này đang mở đường cho cá thể hóa điều trị bổ trợ dựa trên nguy cơ sinh học thực sự của từng bệnh nhân.
Theo dõi động học khối u theo thời gian (longitudinal monitoring)
Khác với cách tiếp cận xét nghiệm đơn lẻ tại một thời điểm, theo dõi động học khối u nhấn mạnh việc đánh giá sự thay đổi của các dòng ung thư theo thời gian. Ví dụ, ở bệnh nhân NSCLC mang đột biến EGFR exon 19 deletion, nồng độ ctDNA ban đầu thường cao trước điều trị TKI, giảm mạnh sau một tháng điều trị, sau đó có thể xuất hiện đột biến T790M ở tháng thứ chín và tăng dần ở tháng thứ mười hai, trong khi hình ảnh học vẫn chưa ghi nhận tiến triển rõ ràng. Trường hợp này minh họa rõ ràng việc tiến triển phân tử xuất hiện trước tiến triển hình ảnh. Theo dõi động học như vậy cho phép dự báo sớm kháng thuốc, chuẩn bị chiến lược điều trị tiếp theo và tránh mất đi thời gian quý giá khi chờ đợi tiến triển hình ảnh.
Phân biệt oligoprogression và systemic progression
Một ứng dụng quan trọng của theo dõi phân tử là phân biệt giữa oligoprogression và systemic progression. Oligoprogression được đặc trưng bởi tiến triển tại số lượng giới hạn vị trí (thường ≤3–5 tổn thương), trong khi phần lớn bệnh vẫn được kiểm soát, thường liên quan đến sự xuất hiện của dòng kháng thuốc mang tính cục bộ. Trong trường hợp này, chiến lược hợp lý là tiếp tục TKI hiện tại kết hợp với điều trị khu trú như xạ trị,đốt sóng cao tần hoặc phẫu thuật. Ngược lại, systemic progression biểu hiện bằng tiến triển đa ổ, ctDNA tăng mạnh và sự lan rộng của các clone kháng thuốc, đòi hỏi phải thay đổi toàn bộ chiến lược điều trị toàn thân.
Từ reactive medicine (y học phản ứng) đến anticipatory oncology (dự đoán ung thư học)
Sự chuyển dịch từ mô hình “đợi bệnh tiến triển rồi mới đổi thuốc” sang mô hình “dự đoán tiến triển và can thiệp sớm” phản ánh sự thay đổi căn bản trong tư duy ung thư học hiện đại. Theo dõi phân tử cho phép bác sĩ đi trước một bước trong cuộc chạy đua với tiến hóa khối u.
Khung chiến lược theo dõi hiện đại
Một chiến lược theo dõi hiện đại có thể bao gồm việc thực hiện NGS trên mô và huyết tương tại thời điểm ban đầu, theo dõi ctDNA định kỳ mỗi 2–3 tháng, và khi phát hiện dòng kháng thuốc, cần đánh giá mức độ, khả năng phối hợp điều trị cũng như phân biệt oligoprogression hay systemic progression. Từ đó, chiến lược điều trị có thể được điều chỉnh trước khi bệnh đạt tiêu chuẩn tiến triển theo RECIST.
6. Multi-omics, trí tuệ nhân tạo và tương lai của điều trị cá thể hóa trong NSCLC đột biến EGFR
6.1. Giới hạn của phân tích đơn tầng gen (single-omics)
Trong nhiều năm, phân tích hệ gen (genomics), đặc biệt là phát hiện đột biến EGFR, đã đóng vai trò trung tâm trong việc định hướng điều trị NSCLC. Tuy nhiên, ngày càng có nhiều bằng chứng cho thấy genomics đơn thuần không đủ để giải thích toàn bộ hiện tượng kháng thuốc và tiến hóa khối u.Nhiều bệnh nhân có biểu hiện kháng thuốc rõ ràng về mặt lâm sàng và phân tử nhưng không phát hiện được đột biến kháng thuốc mới trên DNA, trong khi những thay đổi ở mức phiên mã, biểu sinh hoặc protein lại đóng vai trò quyết định. Điều này phản ánh bản chất đa tầng của sinh học ung thư, trong đó kiểu hình kháng thuốc không chỉ được quyết định bởi trình tự DNA mà còn bởi các cơ chế điều hòa ở nhiều cấp độ khác nhau.
Multi-omics: tiếp cận toàn diện sinh học khối u
Multi-omics là cách tiếp cận tích hợp nhiều tầng dữ liệu sinh học nhằm mô tả toàn diện trạng thái và hành vi của khối u. Trong NSCLC đột biến EGFR, multi-omics thường bao gồm genomics, epigenomics, transcriptomics, proteomics và metabolomics. Phân tích epigenomics giúp làm rõ vai trò của các biến đổi methyl hóa DNA và tái cấu trúc chromatin trong hiện tượng lineage plasticity và chuyển dạng mô học, như chuyển từ NSCLC sang ung thư phổi tế bào nhỏ. Transcriptomics cho phép phát hiện các chương trình phiên mã liên quan đến EMT, stress tín hiệu hoặc né tránh điều trị mà không kèm theo thay đổi gen. Proteomics phản ánh trực tiếp hoạt hóa của các đường truyền tín hiệu, trong khi metabolomics cung cấp thông tin về tái lập trình chuyển hóa – một đặc điểm ngày càng được công nhận trong kháng thuốc điều trị đích. Việc tích hợp các tầng dữ liệu này cho phép nhận diện các cơ chế kháng thuốc “ẩn”, vượt ra ngoài giới hạn của phân tích DNA truyền thống.
Multi-omics và hiện tượng lineage plasticity (khả năng biến đổi dòng tế bào)
Lineage plasticity, gồm chuyển dạng sang kiểu hình tế bào nhỏ hoặc kiểu hình trung gian thần kinh–nội tiết, là một trong những thách thức lớn nhất trong điều trị NSCLC đột biến EGFR. Hiện tượng này thường không đi kèm với các đột biến gen mới rõ ràng, mà chủ yếu được điều khiển bởi các chương trình phiên mã và biểu sinh. Các nghiên cứu multi-omics đã xác định vai trò trung tâm của việc mất chức năng RB1 và TP53 kết hợp với tái lập trình epigenetic, dẫn đến kích hoạt các mạng lưới phiên mã đặc trưng cho ung thư tế bào nhỏ. Những phát hiện này không chỉ giúp giải thích cơ chế sinh học của lineage plasticity mà còn mở ra các hướng tiếp cận điều trị mới, như nhắm vào các yếu tố điều hòa biểu sinh hoặc các con đường duy trì kiểu hình mới.
Vai trò của trí tuệ nhân tạo trong tích hợp và diễn giải dữ liệu
Khối lượng và độ phức tạp của dữ liệu multi-omics vượtxa khả năng phân tích bằng các phương pháp thống kê truyền thống. Trí tuệ nhân tạo (AI), đặc biệt là machine learning và deep learning, đang trở thành công cụ không thể thiếu trong việc tích hợp, diễn giải và khai thác giá trị lâm sàng của dữ liệu đa tầng. AI có thể được sử dụng để xác định các mẫu (patterns) kháng thuốc tiềm ẩn, dự đoán tiến triển bệnh dựa trên động học ctDNA, hoặc phân tầng bệnh nhân theo nguy cơ kháng thuốc trước khi biểu hiện lâm sàng xuất hiện. Ngoài ra, các mô hình AI còn có tiềm năng hỗ trợ lựa chọn chiến lược phối hợp tối ưu cho từng bệnh nhân, dựa trên đặc điểm sinh học riêng biệt của khối u.
Từ ung thư học chính xác tĩnh đến ung thư học chính xác động
Sự kết hợp giữa multi-omics, ctDNA và AI đang thúc đẩy sự chuyển dịch từ ung thư học chính xác tĩnh – dựa trên một xét nghiệm tại thời điểm chẩn đoán – sang ung thư học chính xác động, trong đó chiến lược điều trị được điều chỉnh liên tục dựa trên sự tiến hóa của khối u. Trong mô hình này, quyết định điều trị không còn dựa trên một “đích” duy nhất, mà dựa trên toàn bộ bối cảnh sinh học đang thay đổi của khối u. Theo dõi động học khối u multi-omics cho phép phát hiện sớm sự chuyển dịch sinh học, từ đó can thiệp kịp thời trước khi xuất hiện kháng thuốc lâm sàng.
Thách thức trong triển khai multi-omics và AI vào thực hành lâm sàng
Mặc dù tiềm năng rất lớn, việc áp dụng multi-omics và AI trong lâm sàng vẫn đối mặt với nhiều thách thức. Các vấn đề bao gồm chi phí cao, tiêu chuẩn hóa quy trình lấy mẫu và phân tích, khả năng tiếp cận công nghệ, cũng như khoảng cách giữa kết quả nghiên cứu và ứng dụng thực tế. Ngoài ra, việc diễn giải kết quả multi-omics đòi hỏi sự phối hợp chặt chẽ giữa bác sĩ lâm sàng, nhà sinh học phân tử và chuyên gia dữ liệu.
Tương lai của điều trị cá thể hóa trong NSCLC đột biến EGFR
Cùng các nghiên cứu đang diễn ra về cơ chế kháng EGFR-TKI, những chiến lược mới nhằm vượt qua và trì hoãn kháng thuốc đang xuất hiện, đồng thời đặt ra yêu cầu cao hơn về khả năng dự đoán và quản lý toàn diện tình trạng kháng EGFR-TKIs. Việc phát hiện các con đường kháng thuốc vẫn còn nhiều thách thức, do phần lớn các cơ chế - đặc biệt là các cơ chế kháng với liệu pháp EGFR nhắm trúng đích mới – vẫn chưa được hiểu rõ.
Ví dụ, các nghiên cứu về kháng thuốc với liệu pháp kết hợp kháng thể đặc hiệu EGFR-MET và EGFR-TKI thế hệ 3 trong điều trị đầu tay chưa xác định được các cơ chế kháng mới. Các cách tiếp cận đa-omics và/hoặc nghiên cứu về vi môi trường khối u (TME) có thể giúp phát hiện thêm các cơ chế này.
Các phân tích tích hợp proteomics, genomics, transcriptomics và phosphoproteomics ở NSCLC đột biến EGFR đã xác định được nhiều con đường tín hiệu quan trọng, yếu tố dự đoán mới về sống còn cũng như các marker tiềm năng để phát hiện sớm kháng EGFR-TKI. Ví dụ soluble cadherin-3 (sCDH3) đã được xác định thông qua phân tích tích hợp proteomics và genomics như một marker huyết thanh dự đoán PFS và OS, đồng thời giúp theo dõi sớm đáp ứng với EGFR-TKI. Ngoài ra, các phân tích proteomics, spatial transcriptomics và phân tích phân tử tích hợp cho thấy thiếu hụt ARID1A liên quan đến kháng osimertinib ở NSCLC đột biến EGFR. Tuy nhiên, các nghiên cứu vẫn đang ở giai đoạn sớm. Những phân tích toàn diện hơn về kháng EGFR-TKI vượt ra ngoài mức độ gen là cần thiết, đặc biệt tập trung vào điều hòa phiên mã, biến đổi protein, biểu sinh, chuyển hóa và vi môi trường khối u. Mặc dù DTPs được công nhận là yếu tố trung tâm trong kháng EGFR-TKI, các cơ chế phân tử nền tảng vẫn chưa được làm sáng tỏ hoàn toàn.
Vai trò của AI
Trí tuệ nhân tạo AI đã cho thấy tiềm năng trong việc xác định nhóm bệnh nhân có nguy cơ cao kháng EGFR-TKI. Các mô hình AI như radimoics và hệ thống AI hoàn toàn tự động (FAIS0 có thể tích hợp dữ liệu lâm sàng, hình ảnh và hệ gen để dự đón đáp ứng điều trị và nguy cơ kháng thuốc, từ đố cho phép phân tầng bệnh nhân chính xác hơn. Tuy nhiên, các mô hình AI dựa trên radimoics trong dự đoán hiệu quả EGFR-TKI dù đầy hứa hẹn nhưng còn nhiều hạn chế. Phần lớn nghiên cứu và thuật toán hiện nay được phát triển trên TKI thế hệ 1, làm hạn chế khả năng áp dụng cho bệnh nhân đang điều trị theo chuẩn hiện tại là TKI thế hệ 3. Mặc dù đã có tiến bộ với deep learning toàn bộ phổi,các mô hình này vẫn chủ yếu dựa trên thuật toán truyền thống, với giá trị ngoại suy và khả năng tái lập còn hạn chế trong các quần thể bệnh nhân và bối cảnh lâm sàng khác nhau. Do đó, các hướng nghiên cứu tương lai cần tập trung phát triển thuật toán mới được thiết kế riêng cho EGFR-TKI thế hệ 3 và các phác đồ phối hợp, nhằm nâng cao hiệu năng dự đoán và khả năng ứng dụng lâm sàng của ung thư học chính xác dựa trên AI.
Trong tương lai, điều trị NSCLC đột biến EGFR có khả năng sẽ dựa trên các mô hình dự báo tích hợp, kết hợp dữ liệu lâm sàng, hình ảnh, ctDNA và multi-omics để xây dựng “bản đồ tiến hóa” riêng cho từng bệnh nhân. Cách tiếp cận này không chỉ nhằm kéo dài thời gian sống thêm mà còn tối ưu hóa chất lượng sống, tránh điều trị quá mức và giảm độc tính không cần thiết.
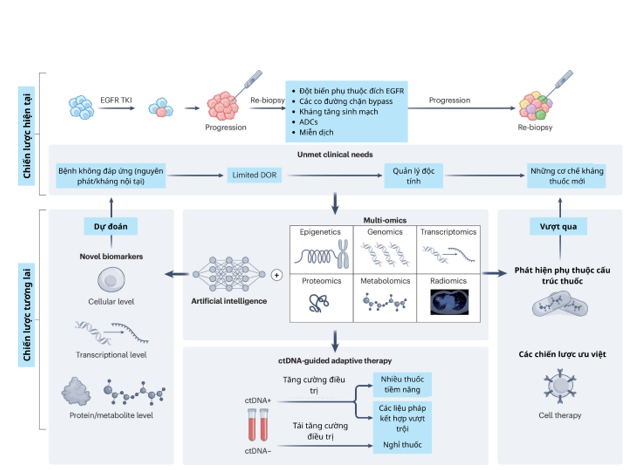
Hình 2 | Chiến lược xử lý tình trạng kháng EGFR-TKI trong NSCLC giai đoạn tiến xa có đột biến EGFR. Phần bảng trên minh họa lộ trình xử lý chung hiện nay với NSCLC đột biến EGFR giai đoạn tiến xa, được điều trị đầu tay bằng TKIs, đồng thời làm nổi bật các nhu cầu lâm sàng chưa được đáp ứng. Phần bảng phía dưới trình bày các chiến lược tương lai trong quản lý tình trạng kháng EGFR-TKI nhằm đáp ứng những nhu cầu lâm sàng này. ADC: antibody-drug conjugate (liên hợp kháng thể-thuốc); ctDNA: circulating tumor DNA (DNA khối u lưu hành); DỎ: duration of response (thời gian đáp ứng).
ctDNA và điều trị thích nghi
ctDNA có thể được sử dụng để theo dõi động học đáp ứng và kháng TKI, từ đó hỗ trợ chiến lược điều trị thích nghi cá thể hóa, gồm tăng cường hoặc giảm cường độ điều trị. Một nghiên cứu tiến cứu đánh giá việc ngưng EGFR-TKI dựa trên ctDNA cho thấy khả năng drug holiday ở bệnh nhân MRD âm tính (thời gian trung vị 9,1 tháng), đồng thời vẫn duy trì hiệu quả khi tái điều trị khi cần (tỉ lệ đáp ứng khi tái điều trị 94%, thời gian trung vị đến điều trị tiếp theo 29,3 tháng).
Thử nghiệm pha III ngẫu nhiên FOCUS-C (NCT05334277) đang tiếp tục đánh giá việc sử dụng sự thanh thải ctDNA như một chỉ dấu để điều chỉnh điều trị, đặc biệt là chiến lược tăng cường điều trị. Cụ thể, bệnh nhân còn tồn tại đột biến EGFR trên ctDNA sau giai đoạn cảm ứng bằng osimertinib sẽ được phân ngẫu nhiên (2:2:1) tiếp tục osimertinib đơn trị, hoặc kết hợp hóa trị platinum kép, hoặc hóa trị kết hợp bevacizumab. Những bệnh nhân đạt ctDNA âm tính sẽ tiếp tục osimertinib đơn trị. Bên cạnh đó, các phương thức điều trị mới nhắm vào các con đường sinh ung thư quan trọng gây kháng EGFR-TKI, như ADCs và kháng thể hai đặc hiệu, đang mở ra những hướng tiếp cận mới để giải quyết thách thức dai dẳng này.
Kết luận Trong hai thập kỷ qua, những tiến bộ quan trọng trong điều trị NSCLC đột biến EGFR đã dần giải quyết các cơ chế kháng thuốc, mang lại hy vọng cho những bệnh nhân trước đây có rất ít lựa chọn điều trị. Tuy nhiên, tế bào ung thư vẫn tiếp tục thích nghi, khiến kháng EGFR-TKI vẫn là một thách thức lâm sàng chưa được giải quyết, đòi hỏi sự làm sáng tỏ sâu hơn cơ chế nền tảng, các chiến lược phát hiện tinh vi hơn và những phương pháp điều trị đổi mới. Mặc dù có nhiều tín hiệu tích cực, vẫn cần nỗ lực nghiên cứu bền bỉ và tăng cường hợp tác toàn cầu để thúc đẩy phát triển các chiến lược điều trị mới, với mục tiêu cuối cùng là cải thiện lâm sàng và lợi ích sống còn cho bệnh nhân NSCLC đột biến EGFR trên toàn thế giới.
⸻ Nguồn: “Navigatingthe landscape of EGFR TKI resistance in EGFR-mutant NSCLC — mechanisms andevolving treatment approaches” trên tạp chí Nature Reviews Clinical Oncology | Tập 23 | Tháng 1 năm 2026 | Trang 63–83)